





When asked to describe his speciality, Kaihang Wang’s answer is immediate: “handyman”. After all, much of his work at the California Institute of Technology in Pasadena involves building things, albeit not with a hammer and nails. Wang and his team develop molecular tools, including a system that biologists can program to introduce a long, synthetic strand of DNA into a bacterial cell1. After further thought, Wang offers more-scientific alternatives: synthetic biology or genome engineering. “All our efforts are fundamentally driven by the goal to make a living thing,” he says.
Like Wang, many biologists reach across disciplines for materials, collaborators or different approaches when the tools at hand fall short. That can lead to methods or consortia that require new descriptors, such as ‘expansion microscopy’ or ‘Genome Project-write’. Some of these create a buzz among scientists through a combination of technical capability and good branding.
Coining a catchy name for a field or tool creates a conceptual infrastructure that researchers can use to frame enquiry, says Erika Szymanski, who studies the rhetoric of science at Colorado State University in Fort Collins. “Just as the constraints of a microscope determine what you can see with it, we can only ‘see’ things when we have names for them,” she says. “Sometimes, trying on a new frame for thinking about the work we do is productive, because it opens up space for imagining new possibilities.”
Here, Nature explores 5 noteworthy technologies from the past 15 years. Some have spawned fields of study or garnered funding; others have increased global collaboration or found fresh purpose in studies far from their original aim. But all have left their mark on science, whether by revealing cell functions, giving rise to companies and therapies or informing public-health policy during a pandemic.
Epitranscriptomics
Like genomic DNA, messenger RNA can carry chemical tags such as methyl or sugar groups that alter its function or fate. Such modifications are not uniform, and the discovery that some mRNAs are highly methylated and others are not hinted at a biological role for the tags. In 2012, RNA biologist Samie Jaffrey at Weill Cornell Medical College in New York City and his colleagues developed a method to identify a specific mRNA methylation mark, named m6A, across the transcriptome (the full complement of RNAs in a cell or organism)2.
Study co-author Christopher Mason, also at Weill Cornell, coined the term epitranscriptomics to explain the team’s hypothesis that the methyl tags regulate the activity of mRNA transcripts, thereby suggesting why protein levels don’t always match the abundances of the transcripts that encode them. “The idea that this might be another layer of the genetic code was very appealing,” Jaffrey says. The new name made it easier for others to grasp the concept.
Over the years, epitranscriptomics has grown into its own field, with specific calls for funding, meetings and collaborations. “In some ways, the fact that a new word was created led to this community,” says RNA biologist Eva Maria Novoa Pardo at the Centre for Genomic Regulation (CRG) in Barcelona, Spain.
Jaffrey and Mason’s original method used an antibody to m6A to isolate fragments of modified RNA that were 100–200 nucleotides long, which they then identified by sequencing. Later, the team cross-linked the antibodies to a substrate, then precipitated the antibody-bound RNA fragments to pinpoint methylated sites, allowing them to generate the first single-nucleotide-level map of methylated mRNA. This helped to identify another class of molecules that carried the modification, called small nucleolar RNAs3. “We’re now starting to coalesce around the idea that a major function of m6A is to mark RNA for quick turnover,” Jaffrey says — which is crucial to a cell’s ability to change and respond to its environment.
Subsequent developments exploited enzymes that could cut non-methylated RNAs at specific sequences. That tool allowed its developer, RNA biologist Schraga Schwartz at the Weizmann Institute of Science in Rehovot, Israel, to detect not just whether a particular site was modified, but also what percentage of transcripts carried the methylated motif. When Schwartz and his colleagues applied this to the entire transcriptome, they found that nearly 75% of modified sites were missed by antibody-based technology, suggesting its sensitivity was limited4. “It was a big surprise to see,” he says. “Having two methods instead of just one allows us to get a more holistic view of the problem.”
Today, epitranscriptomics researchers can read modified RNA directly using nanopore sequencing machines. Unlike conventional sequencers, which require RNA to first be converted to DNA using reverse transcription, these instruments pass RNA molecules through protein nanopores, producing distinctive electrical currents that are then decoded to provide the RNA sequence. Methylated m6A nucleotides are often misread by the sequencing algorithm used to decode the currents. So in 2019, Novoa and her colleagues designed an algorithm (updated earlier this year5) that uses these errors to predict which of those sites carries a methylated nucleotide. “The possibility of sequencing native RNA” — without needing to reverse-transcribe it into DNA first — “ opens up a completely unbiased view of the transcriptome,” she says.
The Human Cell Atlas
The completion of sequencing of the human genome in 2003, together with the arrival of new tools to study single cells, led many to wonder whether they could map every human cell’s unique location, behaviour and development. Sarah Teichmann, a geneticist at the Wellcome Sanger Institute in Hinxton, UK, and Aviv Regev, a computational biologist who is now at Genentech in South San Francisco, California, were among them.
In late 2016, Teichmann, Regev and others convened to discuss the idea. The result was the Human Cell Atlas, a project that uses single-cell approaches to chart the organization, genetics and biology of every human cell, tissue and organ. The group emphasizes an open, collaborative approach: anyone can participate, and the consortium collects information using a wide array of molecular and computational methods.
“There’s no gold-standard technology that can do everything,” says Holger Heyn, who studies single-cell sequencing technologies at the CRG and leads the consortium’s standards and technologies working group. “Every method has biases. The more we integrate diverse technologies, the fewer biases we’ll have.”
In one 2020 study, Heyn and his collaborators compared 13 single-cell RNA sequencing technologies across a common reference set of samples, judging them on their ability to spot cell-specific markers6. One major source of variation in the results, they found, turned out to be the size of cells in a sample. “The goal was not to find a winner or loser, just to define what you might expect to get with each technology,” Heyn says.
The Human Cell Atlas consortium now has almost 2,200 members in 77 countries, who collectively have analysed some 39 million cells from 14 major organs and produced nearly 80 publications, and counting.
Among other things, those data have helped to unlock the mysteries of COVID-19. In early 2020, consortium members pooled 26 published and unpublished data sets to understand how the coronavirus SARS-CoV-2 invades lung tissues. They mapped the cell-surface receptors that the virus uses to enter tissues, including those of the nose, mouth, eyes and more7. Researchers around the world have since used that map to understand the process of infection. It has even helped to inform public-health policies, such as those requiring people to wear face masks, Teichmann says. “The pandemic was really transformative for the Human Cell Atlas project,” she says. “It shows you the value of a cell atlas — even an early, incomplete one.”
Expansion microscopy
Although many researchers obsessed with microscopy resolution have focused on building better hardware, neuroscientist Ed Boyden took a different tack. Together with colleagues at the Massachusetts Institute of Technology in Cambridge, he devised a technique called expansion microscopy, which enlarges cells and tissues like inflating a balloon.
The method infuses a sample with a monomer called acrylate. Adding water causes that monomer to polymerize and swell, pushing cellular components apart as it grows. In early attempts, the cells cracked or swelled unevenly. But adding enzymes to soften tissues before polymerization allowed the researchers to expand mouse brain tissues to 4.5 times their original size8. Two years later, the team extended the method to a dozen tissue types, some of which could be expanded 16-fold9. “Making sure the physical magnification is scaled correctly was essential to making the technique worthwhile,” Boyden says.
This year, Boyden and his team used the concept to locate specific RNAs in tissues, a subfield called spatial transcriptomics. They first expanded a section of mouse brain tissue and then sequenced the embedded RNAs in situ10.
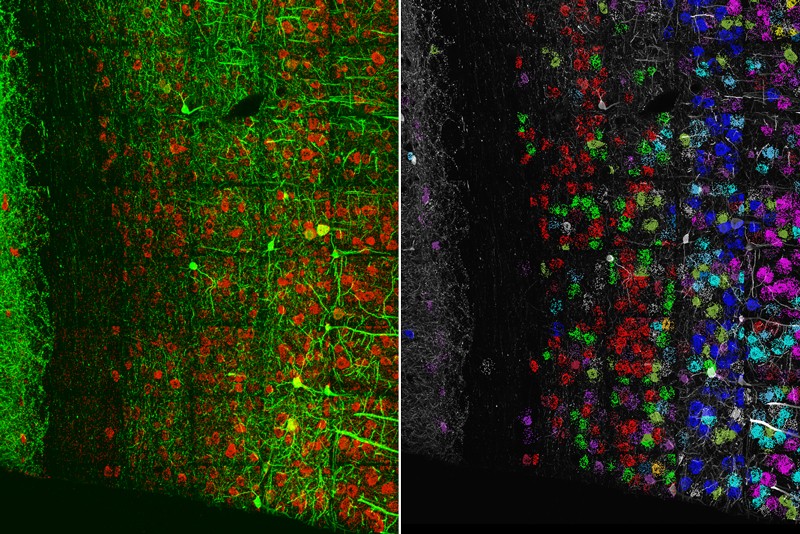
Combining expansion microscopy with RNA sequencing (left) reveals the organization of neurons in the mouse visual cortex (right).Credit: S. Alon et al./Science
Neuroscientist Erin Schuman at the Max Planck Institute for Brain Research in Frankfurt, Germany, who studies how proteins are formed at nerve-cell junctions called synapses, has long relied on indirect methods such as silver staining to visualize this process. Schuman wanted to see newly made proteins in synapses directly. But the synapses are formed by long, thin fibres known as axons that lack good molecular markers. “They’re actually one of the most elusive things to study,” she says.
Using expansion microscopy, Schuman and her team were able to see, for the first time, that almost all axon terminals had the machinery to synthesize new proteins11. “It really helped us access the synapse with a high degree of confidence, and do high-throughput analysis,” she says.
And at Stanford University in California, bioengineer Bo Wang has used the tool to create a high-resolution image of how the common gut pathogen Salmonella interacts with human cells. In optimizing the ‘softening’ step of the process, Wang and his colleagues found that the method could be used to measure the stiffness of the bacterial cell wall. That tough layer is crucial to the pathogen’s resistance to antibiotics and host defences, for instance. Gauging the mechanical properties of microscale objects is difficult, but expansion microscopy helped the team to measure the strength of thousands of cell walls in a single batch, to understand how the bacteria reacted to host-defence mechanisms12. “Similar strategies can help answer physiological questions in plants, fungi and many different species,” Wang says.
Brainbow
In 2007, a team led by neuroscientists Jeff Lichtman and Joshua Sanes at Harvard University in Cambridge, Massachusetts, developed a way to distinguish the tangled skeins of neurons in the mouse brain13. The researchers constructed a system in which genes for a few fluorescent proteins were controlled by regulatory sequences specific to neurons, and flanked by tags that would mark the fluorescent genes to be shuffled in an enzyme-catalysed process called recombination. Cells were given multiple copies of these gene ‘cassettes’, so when the researchers activated a protein that recognized the recombination tags, it shuffled the genes into various, random combinations, expressed as a rainbow of fluorescence. They called their tool Brainbow.
As a graduate student at New York University, Gabriel Victora recalls being awestruck by those kaleidoscopic pictures of the brain, each cell a different hue. But Victora’s studies focused on germinal centres, microstructures in lymph nodes where immune cells divide and grow. “We didn’t immediately think to use this technology,” says Victora, now an immunologist at The Rockefeller University in New York City. “I remember thinking, ‘pity that’s in the brain’.”
Lichtman hoped that the ability to label individual cells would help to resolve fine-scale details such as synaptic connections in the brain. But small cellular structures have fewer fluorescent molecules, making them dimmer — often too dim to be useful. Disappointed with the results, Lichtman says he has since turned to techniques such as serial block-face scanning electron microscopy, in which a block of tissue is repeatedly imaged, peeled back and imaged again to map neural connections. “You have to find the right tool for the job, and in this case, Brainbow wasn’t quite adequate,” he says.
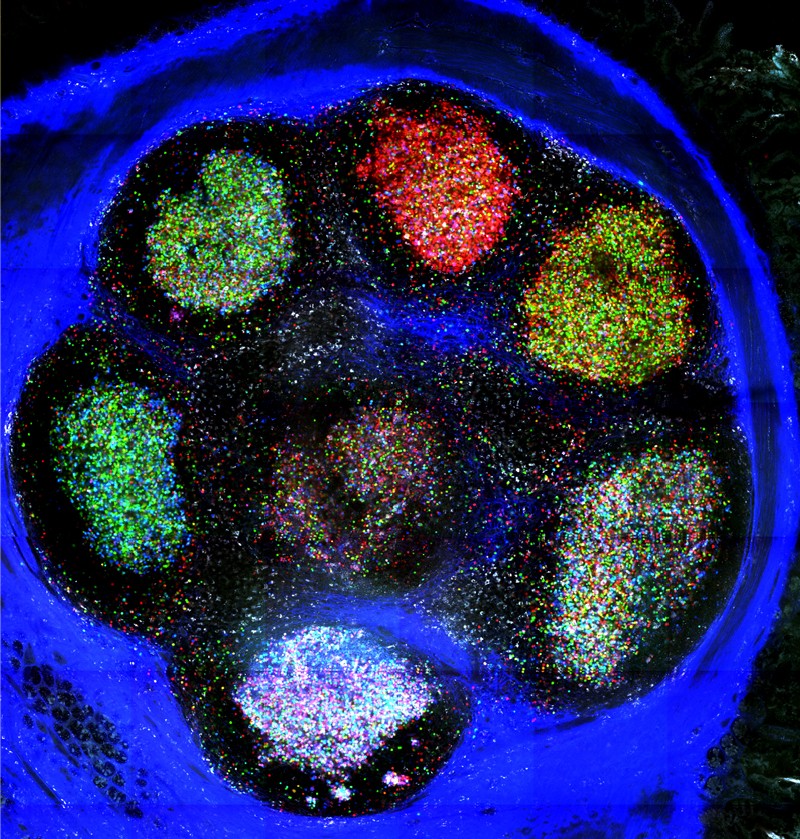
Brainbow-labelled germinal centres.Credit: Carla Nowosad
Lichtman does use Brainbow for experiments in the peripheral nervous system, where cells are farther apart so even dim fluorescence can be observed. And other groups have adapted the tool for different organisms — Flybow for Drosophila brains and Zebrabow for zebrafish tissues, for instance. Combining Brainbow with expansion microscopy has allowed researchers to examine cellular shapes and connectivity in mammalian tissue14.
For Victora, it was a mouse model called Confetti, which extends the technology to non-neuronal cells, that reignited his interest in Brainbow. Inside the germinal centres of lymph nodes, clusters of B cells produce different antibodies and compete to thrive. Most germinal centres maintain a diversity of antibody molecules. But in 5–10% of these structures, Victora and his team found that cells that produce high-affinity antibodies can quickly outcompete other B cells and take over a germinal centre15. Researchers tracking these ‘clonal bursts’ with Brainbow see all the cells in a germinal centre in different colours when they first label cells. Then, as one dominant clone takes over, its progeny — all of which bear the same colour as the parent cell — turn the structure from technicolour to monochrome. “Brainbow shows this division of labour [between B cells] very clearly,” he says.
Genome Project-write
If scientists could make complete synthetic chromosomes, they could confer new functions on cells, swap out disease-causing genetic pathways or design new experimental systems for research. But synthetic chromosomes cannot be built in one go.
In 2010, researchers pieced together the first synthetic bacterial genome16. They remade the organism’s DNA in short chunks, stitched these together, then swapped portions of the chromosome one piece at a time until the native DNA was entirely replaced by its synthetic counterpart. The process has remained largely unchanged since this first attempt, says Wang at the California Institute of Technology. Despite remarkable progress in bacteria and yeast, the technique had never been extended to organisms with more complex genomes. Then, in 2016, researchers announced Genome Project-write, which aimed to synthesize complicated genomes, including that of humans.
Launched with great excitement, the project had to scale back its aspirations — owing to both funding and technical challenges (see Nature 557, 16–17; 2018) — to focus on engineering a human cell line that is resistant to viruses. But DNA synthesis on that scale remains a challenge, as does the design of genetic circuits that encode new functions. For the moment, such work largely remains the purview of individual researchers or small teams, says Christopher Voigt, a synthetic biologist at the Massachusetts Institute of Technology. That process that must shift if larger-scale genome synthesis is to become viable. “It’s something like a single person building an airplane, doing everything from designing it to gluing parts together,” he says. “It shows how far we are from being able to design something at the scale of a genome.”
Still, that lofty goal can spur the field forwards, Wang says. “The motivation to make a whole genome drives the development of technology. It’s a loop: once we have the tools, it makes genome synthesis more realistic and people pour more resources into the field.”
More News
NIH pay raise for postdocs and PhD students could have US ripple effect
Daily briefing: Visual clutter skews our time perception
The Middle East’s largest hypersaline lake risks turning into an environmental disaster zone