




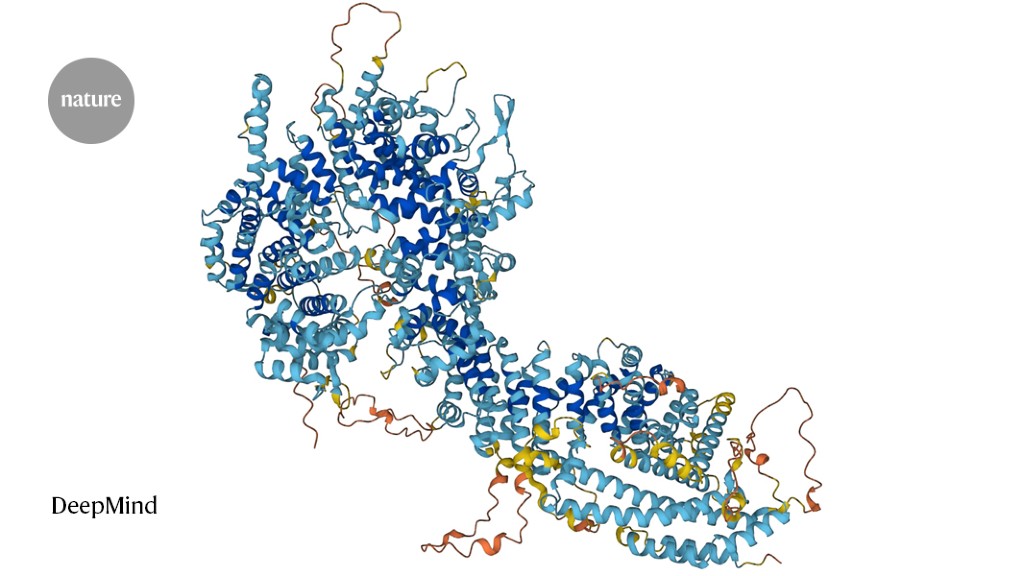
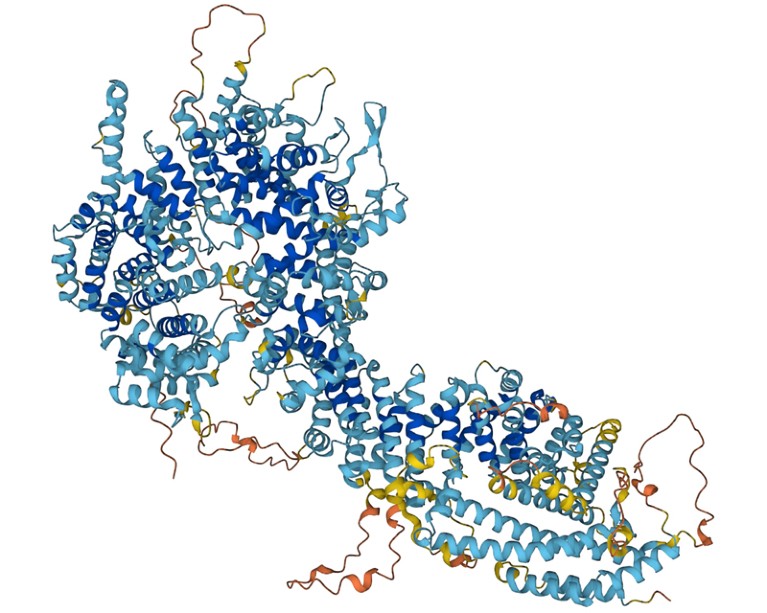
Foldseek allows researchers to identify proteins whose shape resembles that of other proteins.Credit: DeepMind
When you discover a protein, how do you determine what it does? That’s the problem Gregory Gloor was facing.
A biochemist at the University of Western Ontario in London, Canada, Gloor was studying bacterial communities at an oil-refinery wastewater treatment plant, hoping to identify the proteins that help them to degrade toxic substances. As a proof of concept, he started looking at the proteins expressed by viruses called bacteriophages that infect those bacteria. Unfortunately, a search of databases of known proteins for matches came up empty.
Then Gloor learned of a search tool called Foldseek, first shared by its creators in 2021 and described in May in Nature Biotechnology1. “It was like, hallelujah,” he says. His project “went from basically impossible to possible”.
Proteins are built of chains of amino acids, and their folded shape dictates their function. In the past few years, artificial-intelligence tools that predict a protein’s 3D structure from its amino-acid sequence alone — as opposed to determining that structure experimentally — have improved drastically. Researchers have used AlphaFold 2, from Google DeepMind in London; RoseTTAFold, from a team at the University of Washington, Seattle; and other such tools to compile databases containing hundreds of millions of structures. Foldseek makes it possible to quickly search those databases for proteins that have similar shapes — and presumably, similar functions — to a protein of interest.
Best of both worlds
The conventional computational approach to determining the function of an unfamiliar protein is to look for proteins with similar amino-acid sequences. If the functions of those related proteins are known, researchers can make a guess as to what the new protein might do.
Sequence searches are fast, like searching a hard drive for a file name. But they often miss good matches because proteins with similar shapes can have vastly different sequences. Structure-based search methods look for shapes instead of sequences, but this can take thousands of times longer, because it’s computationally difficult to compare complex 3D objects. With Foldseek, researchers got the best of both worlds: the software represents a protein’s shape as a string of letters — a ‘structural alphabet’ — thereby offering the sensitivity of shape-based searches but at the speed of sequence-based ones.
“One of the key ideas was that in order to produce a good structural search, it is important to get the encoding right,” says Martin Steinegger, a biologist at Seoul National University and one of the Foldseek paper’s lead authors.

What’s next for AlphaFold and the AI protein-folding revolution
Gloor used ColabFold, a cloud-based computational-notebook interface to AlphaFold 2, to predict the structures of the bacteriophage proteins he found, and then Foldseek to match them to known proteins. Some of the proteins, he found, formed the viruses’ outer shells; others were enzymes2. His assessment: Foldseek is “amazingly clever”.
Foldseek is not the first algorithm to reduce protein structure to an alphabet. Other search tools typically assign each amino acid a letter on the basis of its orientation relative to the amino acids immediately before and after it in the protein sequence. However, that approach overlooks interactions between amino acids that are far apart in the linear chain, but nearby in 3D space. Foldseek assigns each amino acid one of 20 letters, on the basis of its distance from, and orientation relative to, the amino acid that’s closest in the folded-up protein. By focusing on these spatial bridges, Steinegger says, Foldseek’s ‘3D-interaction alphabet’ better captures global structure.
Seeing back in time
“Biology occurs in three dimensions,” says Janet Thornton, a computational biologist at the European Molecular Biology Laboratory’s European Bioinformatics Institute in Hinxton, UK. The ability to compare proteins on the basis of their shape “allows you to see much farther back in evolutionary time, which allows you to identify very distant relatives that evolved from the same precursor” protein, she says.
To test Foldseek, Steinegger’s team used a database of 365,000 proteins whose shapes had been predicted using AlphaFold 2. They fed 100 of these shapes into Foldseek and asked it to rank, for each one, the most similar proteins in the database. The score was based on how many ‘true positives’ the algorithm retrieved (that is, proteins scoring above a certain similarity threshold according to atomic modelling) before retrieving a false positive. Foldseek outperformed two popular structure-based search tools, TM-align and Dali — performing 24% and 8% better, respectively — and nearly 35,000 and 20,000 faster. Compared with a structural-alphabet-based tool called CLE-SW, Foldseek was 23% better, and 11 times as fast1.
DeepMind’s AI predicts structures for a vast trove of proteins
Foldseek is available as open-source software for macOS and Linux computers. The developers also created a web server for researchers to search any of seven structural databases covering hundreds of millions of proteins. According to Steinegger, the software has been installed at least 14,000 times, and researchers run about 300 searches on the server each day.
Thornton says Foldseek could help researchers to identify protein functions in new pathogens, or simply shed light on how organisms operate. For example, Steinegger and his team applied Foldseek to find clusters of related proteins in the AlphaFold database and identified bacterial proteins with a similar structure to a human histone3.
As for Gloor, with existing search tools, he found matches for only a small fraction of the bacteriophage proteins in his study, none of which had known functions. Using Foldseek, he found matches for half of his proteins, identifying 15% as enzymes2.
“Converting a three-dimensional volume of interactions down into a string required a fair bit of insight and originality,” Gloor says. And using Foldseek, scientists can understand many more proteins in many more organisms. “It’s really going to change the way that we do evolutionary studies,” he says. “It will increase our ability to look in truly unique ecosystems and figure out how they work.”
More News
Author Correction: Bitter taste receptor activation by cholesterol and an intracellular tastant – Nature
Audio long read: How does ChatGPT ‘think’? Psychology and neuroscience crack open AI large language models
Ozempic keeps wowing: trial data show benefits for kidney disease