




The road to gene therapies for genetic disorders has been long — and expensive — but the field could soon get some good news. On 27 June, the US Food and Drug Administration will decide whether to grant a fast-track approval to the first gene therapy for Duchenne muscular dystrophy (DMD), a genetic disorder that affects around 1 in 3,500 boys. Children with DMD can’t make a protein called dystrophin, resulting in progressive muscle degeneration and death in their twenties due to heart or respiratory failure.
The therapy, known as SRP-9001, is made by Sarepta Therapeutics based in Cambridge, Massachusetts. It would be the 13th gene therapy that the FDA has approved since 2017, and the first to target a prevalent genetic disease in children. The accelerated approval would allow the drug to reach the market before large clinical trials have been completed, on the basis of evidence that the therapy allows boys to make an engineered form of dystrophin.
Gene therapy’s comeback: how scientists are trying to make it safer
The decision date was delayed late in May after FDA officials and advisers raised concerns about the strength of Sarepta’s data so far; SRP-9001 seems to have only a modest effect on muscle function, and only in some people. A press release from the company says that the agency is likely to approve it only for boys aged four and five, but might expand that range if an ongoing clinical trial shows that the drug’s efficacy is worth the risks associated with it. Analysts predict that the one-time treatment will cost US$2 million. A Sarepta spokesperson declined to reveal the price tag until the drug is approved but says that it will be priced “below the value it will bring to patients”.
Still, some scientists hope that the approval will pave the way for more gene therapies. “To have a big success like this is going to validate the technology quite a bit,” says Jeffrey Chamberlain, a neurologist at the University of Washington in Seattle who developed some of the technologies used by Sarepta. “It’s a way to get our foot in the door now and help kids.”
Why has DMD been so challenging to treat?
DMD seems like a straightforward target for gene therapy. The disease affects boys almost exclusively, because they have only one copy of the X chromosome, where the dystrophin gene is located; girls with a disease-causing variant have a backup copy. Replacing even some working protein in muscle cells should reverse the disease, or at least halt its progression.
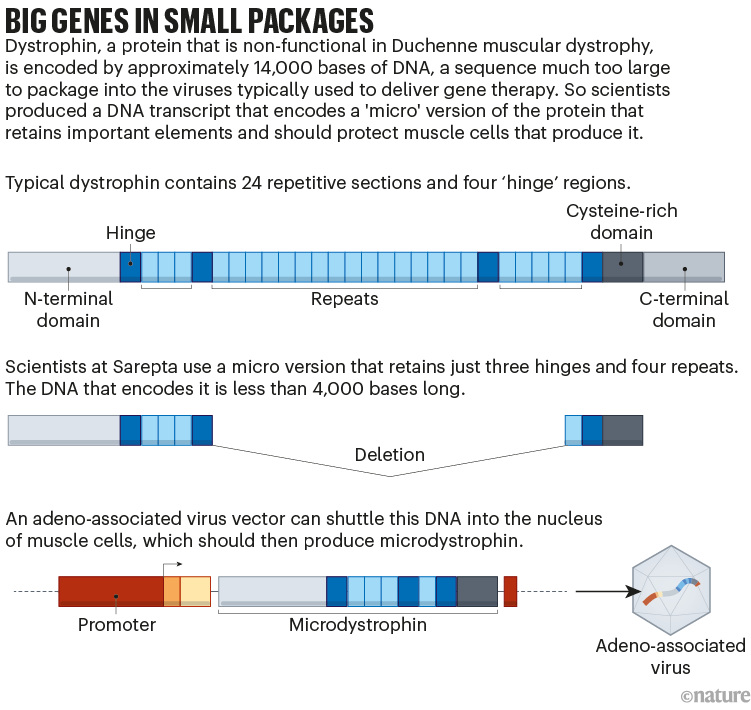
But developing that replacement has proved difficult. Dystrophin is the longest gene in the human genome and is much too large to fit into the adeno-associated virus (AAV) vector commonly used to deliver gene therapies. Sarepta and several other companies have got around this by designing a gene that encodes just the most important parts of the protein (see ‘Big genes in small packages’). The resulting ‘microdystrophin’ is only partly effective.
Getting the gene into enough cells to make a difference has also been challenging. Muscle makes up 30–40% of the body’s mass, so treating DMD requires extremely high doses of the AAV vector. This raises the risk of side effects, including organ damage. And because muscle cells divide frequently — especially in a growing child — the amount of the gene in the body will be diluted over time. It’s still unclear how long the therapy’s effect will last, although Sarepta’s data from four children suggest that the clinical improvements can persist four years after treatment.
How effective is the treatment?
Although most FDA approvals require drugs to pass two phase III, placebo-controlled clinical trials testing the efficacy of the treatment, the accelerated approval pathway that Sarepta is seeking allows companies to rely on “surrogate endpoints” to prove their value. These measures show a biological effect — in this case, high levels of microdystrophin — rather than an improvement in symptoms.
Sarepta’s first large phase III trial is still under way, with data expected to be released late this year. The FDA has instead evaluated several midstage trials in a total of 52 boys aged four to seven. These showed that the boys’ muscles made microdystrophin, but the children had no statistically significant improvement in muscle function one year after the drug was administered.

Will the FDA change how it vets drugs following the Alzheimer’s debacle?
Reporting by the news organization STAT and others found that FDA staff planned to reject Sarepta’s application. But Peter Marks, director of the FDA’s Center for Biologics Evaluation and Research (CBER) intervened — scheduling a public meeting with an independent scientific advisory committee on 12 May, to weigh in on whether the drug should receive an accelerated approval. This committee narrowly recommended the approval by eight votes to six. Although the FDA doesn’t have to follow the advice of the committee, it typically does.
The advisory committee evaluated evidence for surrogate markers as well for clinical benefits. Some of those who voted in favour of the therapy say that because DMD progresses slowly, it might be difficult to determine the drug’s effect after just one year. But when Sarepta looked only at the 32 boys younger than six years old who received SRP-9001 or a placebo, the treated boys’ muscle function was significantly improved.
Sarepta’s chief scientific officer, Louise Rodino-Klapac, doesn’t think that the drug actually functions better in these younger boys. Rather, she says, the older boys in the placebo control group were healthier at the beginning of the study than their younger counterparts, making the drug’s effects look weaker in this group by comparison. Rodino-Klapac expects that the ongoing phase III trial, which better controlled for each patient’s initial condition, will show that it works equally well in older boys’ muscles.
The statistically significant improvement in the younger boys was enough to convince Donald Kohn, an FDA advisory committee member who does stem-cell research at the University of California Los Angeles, to vote in favour of the drug. “From a statistical standpoint, [Sarepta] haven’t pled their case,” he says. But, he adds, the efficacy in younger boys suggests that the drug is having a real effect. “It’s a vote for hope,” says Kohn.
Others are more concerned by what looks like an overall lack of efficacy. “I understand the appeal of ‘Something is better than nothing’, but the question is at what cost,” says Caleb Alexander, an epidemiologist at Johns Hopkins University in Baltimore who was also a committee member and voted against the drug. Not only can SRP-9001 have significant side effects, he says, but the placebo-controlled trials completed so far do not strongly suggest that high levels of microdystrophin even predict clinical outcomes. “No number of post-hoc analyses of subgroups of boys change that fact,” he says.
The FDA and advisory committee members also raised concerns about whether Sarepta would finish its clinical trial, which is still evaluating more than 60 boys who had initially received the placebo and are now receiving the drug. They pointed out that the company did not complete trials of three previous DMD drugs that the FDA had approved on an accelerated basis. They are still on the market. A spokesperson for Sarepta says that those trials have now enrolled enough patients and are now under way.
Expensive treatments for genetic disorders are arriving. But who should foot the bill?
“It’s a really complex decision. I don’t envy the FDA,” says Sharon Hesterlee, chief research officer of the Muscular Dystrophy Association in Chicago. Although she agrees that the therapy’s effect might be modest and carries significant risks, “the risk of doing nothing is 100% fatal”.
What are the risks of DMD gene therapy?
As well as the risks from the large amounts of AAV required to get the microdystrophin gene into muscles, the gene itself appears to cause severe side effects in some children. The FDA had suspended multiple DMD gene therapy trials owing to contamination concerns and patients becoming seriously ill. And in 2021, the agency temporarily halted a study by Pfizer, based in New York City, which is testing its own version of microdystrophin, after a patient died.
To investigate the problem, Pfizer, Sarepta and Solid Biosciences, based in Charlestown, Massachusetts, decided to pool their microdystrophin data. Their study, which will soon be published in the New England Journal of Medicine, found that certain mutations in the dystrophin gene cause the immune system to recognize microdystrophin as a foreign invader and attack it, causing dangerous inflammation in the muscle and heart. The companies began genetically screening clinical trial participants and excluding those with the relevant mutations — less than 5% in Sarepta’s case — which eliminates the problem for now. But Carsten Bönnemann, a neurologist at the National Institute of Neurological Disorders and Stroke in Bethesda, Maryland, who co-led the study, says that gene therapies will eventually have to be tailored to help these patients.
Gene therapies delivered by adeno-associated viruses can only be administered once in a patient.Credit: Mehau Kulyk/Science Photo Library
Still, Hesterlee notes that even those who are eligible for Sarepta’s newly approved therapy have a tough choice ahead of them. AAV gene therapies can be administered only one time in a person’s life: once the immune system has encountered a virus vector, it is likely to attack it in the future. And because AAV is the vector of choice for most gene therapies, parents of a child with DMD might have to choose whether to treat their child now, with the only approved therapy, or wait in the hope that Sarepta or another company will release something better in the future. That means allowing the disease to progress, and potentially stabilizing it only after boys have lost more muscle function. “A few months can be a lot for families,” Hesterlee says.
What other DMD gene therapies are on the horizon?
Pfizer is conducting a phase III trial in 99 boys with their version of microdystrophin in a similar AAV vector and expects to release its initial results next year. Several other companies are developing their own microdystrophins, including Regenxbio in Rockville, Maryland, which launched a small trial earlier this year.
Others are searching for ways to improve the viral vector that carries the gene. In 2022, Solid Biosciences halted its phase II trial of its own microdystrophin formulation to switch to a different vector that specifically targets muscle cells and can probably be given in lower doses. The company plans to begin dosing patients later this year. And at the American Society for Gene & Cell Therapy’s annual meeting in Los Angeles in May, Chamberlain presented a new system involving several AAV vectors that each carry a portion of the dystrophin gene. The gene products can then link together to create a version of dystrophin that is much larger than a single vector could carry. Further on the horizon are approaches that will infuse patients with genes encoding the CRISPR/Cas9 genome editing system with the intention of permanently altering the dystrophin gene itself in muscle cells.
Researchers welcome $3.5-million haemophilia gene therapy — but questions remain
And other researchers are looking into alternative vectors, including lipid capsules, nanoparticles, antibodies that carry dystrophin genes to muscle cells and viruses that integrate into a patient’s genome. These treatments, at least in theory, could be administered to people who have already received AAV gene therapy.
But with an approved therapy now available, companies trying to test their own therapies could find it more difficult to recruit trial participants, who might not want to risk getting a placebo. Furthermore, health-services researcher Reshma Ramachandran at Yale University in New Haven, Connecticut, worries that if companies know that they can get a microdystrophin therapy approved, they will be more likely to create their own slightly tweaked versions instead of spending a longer time developing innovative approaches of their own.
How will this approval affect other gene therapies?
Gene-therapy development has always been a risky and expensive business, and the recent economic downturn has seen many biotechs shut down. Nevertheless, the number of gene-therapy approvals by the FDA has risen each year since its first one — Kymriah for leukemia — in 2017.
Approval for a DMD gene therapy would still represent a milestone, however. Until now, most approved gene therapies have addressed cancers, extremely rare diseases and conditions such as retinal disorders, which are easy to target with a virus. DMD is different, Chamberlain says, both in the number of technical challenges that researchers had to address and in the large number of patients that the therapy could serve. The agency is now poised to consider more than a dozen gene and cell therapies this year, including two for sickle-cell disease — a genetic condition that is much more prevalent than DMD.
The FDA seems to be prioritizing these approaches. In recent talks, CBER director Marks said that the agency plans to lean more heavily on accelerated approval pathways for gene therapies, including broader use of surrogate endpoints. “We can’t be so careful about our approvals under accelerated approval that we prevent potentially life-saving therapies from getting to market in a timely manner,” he said in a January speech at the Muscular Dystrophy Association’s annual meeting.
That concerns some scientists, who worry that patients will end up paying high prices for ineffective drugs. “I do think the notion of regulatory flexibility can get bent out of shape,” Alexander says. Ramachandran points to the FDA’s 2021 accelerated approval of an Alzheimer’s drug, aducanumab, that successfully eliminated amyloid plaques in patients’ brains but did not measurably improve their health. The FDA subsequently approved another, similar drug that showed only minor clinical benefit. “Patients with Duchenne’s deserve not just more options, they need better options,” Ramachandran says.
But Chamberlain contends that even a small amount of success will help the whole field. “There’s tremendous room for improvement,” he says. “I think this is going to buy us some time to make those improvements.”
More News
Judge dismisses superconductivity physicist’s lawsuit against university
Future of Humanity Institute shuts: what’s next for ‘deep future’ research?
Star Formation Shut Down by Multiphase Gas Outflow in a Galaxy at a Redshift of 2.45 – Nature