





Credit: Taj Francis
Sickle-cell disease was once a childhood ailment, simply because many children with the condition died before reaching adulthood. But, since 1972, when the US National Sickle Cell Disease Program was founded and specific funding became available, the United States has managed to drastically reduce childhood deaths from the disease. Between 1979 and 2005, the death rate for children with the disorder dropped by an average of 3% per year1.
Unfortunately, although the outlook has improved for children, it has worsened for adults. In all the time that researchers have focused on childhood sickle-cell disease, “there wasn’t as much attention on the fact that these children become adults”, says Marsha Treadwell, a clinical psychologist and sickle-cell specialist at the University of California San Francisco (UCSF) Benioff Children’s Hospital in Oakland. The death rate for adults with sickle cell in the United States increased by 1% each year between 1979 and 20051, and this trend continued until at least 20172.
Access to paediatric care is far from perfect — only one-third of children with sickle-cell disease are adequately checked for their risk of stroke3, for example. In the United States, most people with the disease — although certainly not all — are Black or Latinx with African ancestry. Systemic racism throughout the country’s history has resulted in these populations having less wealth, lower social status, fewer job opportunities and poorer access to health care than the white majority. This means that sickle-cell care has been plagued by inequities: people with the disease are less likely to have private health insurance or disposable income, so treating them is less profitable, leading to less research funding and not enough specialists.
But adult care lags behind even more. The difference cannot be fully explained by biology — many of the interventions that save children’s lives can save adult lives, too, but they are rarely used. Instead, it is underpinned by a lack of health-care resources devoted to adults with sickle cell, plus a stronger influence of racism when adults interact with the medical system.
Paediatric care has improved so much over the past decades that it provides a road map for adult care. “Adult sickle-cell programmes should be funded like the paediatric ones are,” says Sophie Lanzkron, a haematologist specializing in adults at the Johns Hopkins University School of Medicine in Baltimore, Maryland. It’s not about education: specialists generally know what to do, the health-care system just isn’t doing it, she says.
It takes a team
The biggest reason adult care isn’t in line with paediatric care is that there are few comprehensive centres for adults with sickle-cell. “The model of comprehensive care is the model that really resulted in great advances for children so that they could live longer,” says Treadwell. The National Sickle Cell Anemia Control Act of 1972 allocated funding for sickle-cell screening, treatment, research and education, and it spurred the creation of multidisciplinary care centres for children across the United States. The National Institutes of Health granted funding for 10 centres in 1972 and 5 more in 1973, with extra grants throughout the subsequent decades — the latest being in 2008. These centres included not only haematologists who specialized in sickle cell and emergency-department physicians well versed in the disease, but also social workers, psychologists, education specialists and nutritionists. Specialists in treating organs commonly damaged by sickling cells, especially the brain and kidneys, could also be looped in.
But adults with sickle cell do not typically have access to such centres. There are only 49 adult sickle-cell centres in the United States versus 168 for children, according to the Sickle Cell Disease Association of America. That’s compared with more than 100 centres each for cystic fibrosis and haemophilia, diseases of similar complexity and frequency, which affect a significantly greater number of white people.
Because of this lack of facilities, says Treadwell, “there was a push to move people with sickle-cell disease into primary care — but that’s not really appropriate for a multi-system, complex, chronic condition.” This is “an example of structural racism”, she says. “With cystic fibrosis, it’s a no-brainer that the specialist directs the cystic fibrosis care. In cancer, it’s a no-brainer that the oncologist directs the cancer care.” But with sickle cell — a disease that disproportionately affects people with African ancestry — individuals are left to do the best they can with general practitioners.
And that, sickle-cell specialists say, is a recipe for failure. Not only is specialized knowledge required to treat people properly, but the disease is also just uncommon enough that most primary-care physicians can’t afford to spend their time keeping up on it. They might only have one person with sickle cell and 100 with diabetes. “You’re going to spend your continuing medical education on figuring out how to best treat diabetes and not on sickle-cell disease,” says Lanzkron.
Filling the gaps
So, comprehensive-care centres are a must. Fortunately, there are ways to catch up on building them for adults. John Strouse, a haematologist who treats adults and children at Duke University School of Medicine in Durham, North Carolina, is impressed by California’s approach. Last year, the US Centers for Disease Control and Prevention (CDC) in Atlanta, Georgia, funded statewide surveillance programmes to collect data on the prevalence and distribution of sickle-cell disease. “California leveraged that data to get US$15 million from their state legislature to start sickle-cell centres,” Strouse says, “an amount of money that is potentially transformative.”
Treadwell was one of the leaders of the California effort. The group met every month for a year, she says, to craft a plan for the state legislature. So far, ten sickle-cell centres have been funded, including some in southern California — the part of the state where adult sickle cell is most prevalent, she says.
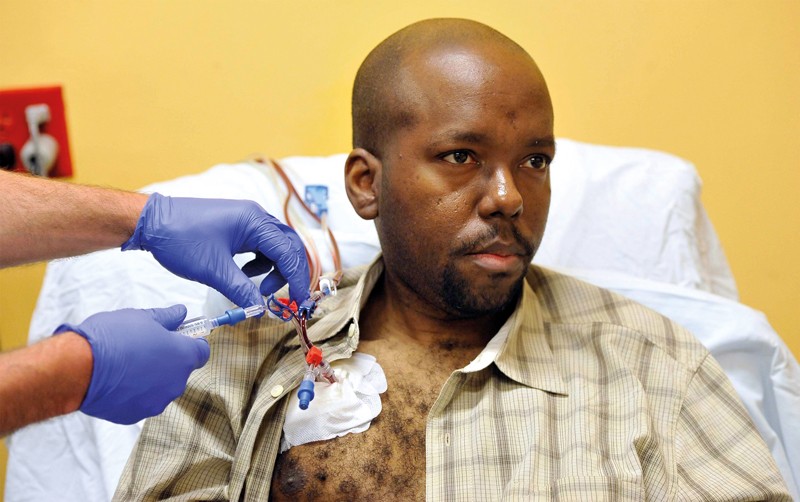
A person with sickle-cell disease receives a blood transfusion in a hospital in Maryland.Credit: Baltimore Sun/TNS/Getty
In places with no specialized centres, a hospital should instead direct people with sickle-cell disease to teams in the hospital that care for people with complex medical needs, such as those with mental illness as well as congestive heart failure, says Jeffrey Glassberg, director of the Mount Sinai Comprehensive Sickle Cell Program in New York City. For rural or remote medical systems that are too small to do that, Lanzkron hosts a weekly, hour-long telementoring meeting. Remote medical providers call in to present and discuss cases with Lanzkron and a team of sickle-cell specialists, including a psychiatrist who is also a pain expert, and often social workers and community health workers. Telementoring flips the script on telehealth: people get in-person care from a generalist, who in turn gets virtual support and advice from sickle-cell specialists.
Because of the lack of specialized disease centres for adults, the switch from paediatric to adult care — often referred to by caregivers as transition — is particularly difficult. “I hate transition,” Lanzkron says. Paediatric centres typically have not only specific sickle-cell specialists, but also social workers and patient-care coordinators to remind individuals of their appointments. That can be crucial for people with sickle cell, who can be struck by silent strokes that can interfere with their ability to make appointments. “The minute you hit transition age, there’s nothing,” says Lanzkron. Perhaps as a result, pain episodes and organ damage become increasingly common around the age of 18 — more so than would be expected simply from increased age, says Kim Smith-Whitley, a paediatric haematologist at Global Blood Therapeutics, a pharmaceutical company in San Francisco, California, that focuses on sickle-cell disease.
Preventive treatment
The past several decades have also seen a push in paediatrics to prescribe hydroxyurea — a life-saving preventive treatment that also works for adults3. Originally developed as a chemotherapy drug for cancer, hydroxyurea is now the recommended treatment for managing the side effects of sickle cell4.
Hydroxyurea reduces both the frequency and intensity of the severe pain crises in sickle-cell disease4, but needs to be given at the right dose for each person. It can be prescribed as early as nine months of age, says Smith-Whitley. It is available as a generic drug and is not terribly expensive, costing typically about $1,000 per year per person. “Unequivocally, it works,” says Glassberg. “It makes people healthier, it makes people live longer.” It’s no secret, and yet only about 30% of adults with sickle cell take it. Glassberg says that about 80% should be using this medication — it does not work in people with the haemoglobin SC genotype of sickle cell, and should not be given to people who are pregnant.
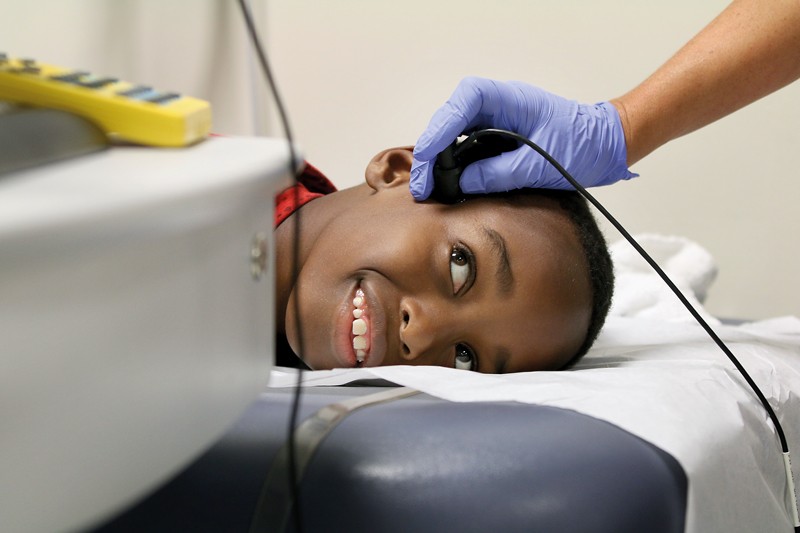
A child gets a transcranial Doppler scan.Credit: Sarah Pack of MUSC
One challenge is that hydroxyurea is not a quick fix. It reduces acute severe pain crises, but a patient might have only two crises each year, says Strouse, so “we recommend trials of a year”. That’s a long time to take a pill every day without concrete evidence that it’s doing anything; unsurprisingly, many people stop the regimen prematurely. Compounding the problem, many non-specialists prescribe too little hydroxyurea for it to be effective. “Haematologist–oncologists that don’t treat many people with sickle-cell disease often use inadequate doses,” Strouse says. Glassberg is part of a team studying whether a mobile app can improve dosing and get people and providers to persist long enough to see benefits.
The other preventive measure that has shown success in paediatric sickle-cell disease is stroke screening. Without screening, 10% of children with sickle cell will have a stroke before the age of 205. Stroke screening can reduce that to just 1–3%. The screening uses a kind of ultrasound called transcranial Doppler (TCD) to measure the blood flow in large blood vessels in the head and neck. Children whose blood flow is faster than normal are identified as high risk. When sickle-shaped red blood cells get trapped in blood vessels in the brain, the vessels narrow, causing faster blood flow and increasing the chance that the next obstruction could lead to a complete blockage and cause a stroke. To lower their risk of stroke, children identified by the screen are given blood transfusions, typically followed by hydroxyurea six months to a year later, says Strouse. Unfortunately, TCD does not work in adults, so they don’t get this risk-assessment benefit, says Lanzkron.
The racism factor
It’s clear that structural racism leads to disparities in resources. But racism also leads to differing treatment in medical care, especially for a disease that causes acute pain crises that are best treated with opioids. When Black people show up at an emergency department in a severe pain crisis, they are often — in large part because of their race — assumed to be drug-seekers. Their pain is discounted.
There are no outside markers of sickle-cell pain. “I can’t do a blood test,” says Lanzkron. “The gold standard is that the patient just told me that they’re having a crisis.” And evidence shows that physicians are more likely to dismiss Black people’s pain. Although anti-Black prejudice applies to all ages, it is much more common against adolescents and adults, who, unlike children, are routinely suspected of seeking opioid pain medication for an addiction.
Opioids are the best treatment for an acute sickle-cell crisis, but providers in emergency departments often don’t know that. They also usually don’t know the patient or have access to background information about how their primary physician is managing their condition.
One of the people that Lanzkron treats went to a different hospital when in an acute pain crisis, and the providers would not accept that she was still in pain after a dose of pain medication, even though this person was herself a nurse. “They told her she had to leave. She said, ‘I’m in horrible pain, I can’t leave.’” recalls Lanzkron. “They called security. They put her in a wheelchair. They wheeled her to the kerb, and her husband brought her to us.” The team then found that she had acute chest syndrome, one of the leading causes of death from sickle-cell disease.
Most of the people that Lanzkron treats have had similar experiences. “My patients who are mothers, my patients who are lawyers and doctors, can all describe episodes of being treated like a drug addict,” she says.
One obvious way to prevent discrimination in hospital is to keep people out of them in the first place. For instance, Strouse says the Duke University hospital system where he works treats about one-third of its people with sickle cell in a day hospital, where they can receive intravenous opiates for pain crises, along with anti-nausea and anti-itching medications. “It’s faster and better than the emergency department,” he says, “and it’s lower cost.” However, it has been difficult for uninsured patients to get day-hospital care, although they can still be seen in the emergency department, he says. Lanzkron is studying a similar model of dedicated sickle-cell infusion clinics.
But no matter what other facilities are made available, people with sickle-cell disease will still end up in hospital. To improve the care they receive when they get there, Glassberg has found success since working at Mount Sinai by tweaking electronic medical records to nudge providers in the correct direction. He and his colleagues added a scrolling ribbon that displays when a person has the disease. The US Agency for Healthcare Research and Quality recommends classifying all people with sickle-cell disease at the second-highest urgency level, meaning “in danger and in need of immediate attention, but not likely to die imminently”, he says. “That got our appropriate triage from 30% to 85%.”
Having a dedicated system for people with sickle cell in an emergency department can also work. When Lanzkron first arrived at Johns Hopkins, the time it took to give the first dose of pain medication to a person with sickle-cell disease was a whopping six hours. The hospital was able to reduce that to about 2.5 hours by opening an observation unit in the department, staffing it with nurse practitioners and physician assistants with sickle-cell expertise and giving priority for those beds to people with sickle cell. A physician’s assistant took it on herself to develop a programme to treat people with sickle cell in the waiting room if necessary, which shaved another hour off of the time to receiving pain medication. Before COVID-19 increased these wait times, “it was a well-oiled machine”, says Lanzkron.
The recent and impending development of drugs for this disease is exciting to sickle-cell disease specialists. “There’s just kind of this buzz and energy in sickle cell,” says Glassberg. He used to go to American Society of Hematology conferences to present on sickle-cell disease, “and you’d be alone in the poster hall”, he says. “Now, layperson investors will tap me on the shoulder and ask me things. Now it’s crowded in the poster halls. Now, we have to focus that energy.” Because the bottom line is that any new pharmaceuticals will need to be backed up by a medical infrastructure that is still severely lacking.
More News
Author Correction: Bitter taste receptor activation by cholesterol and an intracellular tastant – Nature
Audio long read: How does ChatGPT ‘think’? Psychology and neuroscience crack open AI large language models
Ozempic keeps wowing: trial data show benefits for kidney disease